Research projects
Overview
As a scientific researcher in biology, my goal is to improve the understanding of life, in particular at the cellular and molecular level. To do so, I design, develop and use computational methods and tools to analyze genomics, transcriptomics and other -omics data, mostly obtained from sequencing experiments. My field of expertise is called computational biology, it lies at the interface between molecular biology and bioinformatics.
More precisely, my research activities focus on genome annotation and 3D genomics, which mostly involve omics data analysis.
Below are listed a few research projects I am or was involved in.
GENE-SWitCH
 The regulatory GENomE of SWine and CHicken: functional annotation during development
The regulatory GENomE of SWine and CHicken: functional annotation during development
Project description:
GENE-SWitCH aims to deliver new underpinning knowledge on the functional genomes of two main monogastric farm species (pig and chicken) and to enable immediate translation to the pig and poultry sectors. The activation status of functional genome sequences varies across time and space, and in response to environmental perturbations. In full coordination and synergy with global effort and ongoing projects of the Functional Annotation of Animal Genomes (FAANG) community, we will characterize the dynamics (“switches”) of the functional genome from embryo (chicken) and fetus (pig) to adult life by targeting a panel of tissues relevant to sustainable production.
Founding:
EU, 2019-2023
Project status:
Ongoing
Main collaborators:
INRAE/INSERM, Roslin Institute, WUR, EBI
Role in this project:
WP2 contributor
FR-AgENCODE
 FR-AgENCODE: a FAANG pilot project for the functional annotation of livestock genomes
FR-AgENCODE: a FAANG pilot project for the functional annotation of livestock genomes
Project description:
As part of the FAANG action (Functional Annotation of ANimal Genomes), the FR-AgENCODE project aims at improving the genomic annotation of 4 livestock species: cattle (Bos taurus), goat (Capra hircus), chicken (Gallus gallus), pig (Sus scrofa). This is achieved by performing molecular assays on tissue dissociated cells (liver) and on sorted primary cells (CD4+ and CD8+ T lymphocytes) from 2 males and 2 females of each species. These assays include RNA-seq, ATAC-seq and Hi-C to characterize the transcriptome, the chromatin accessibility and the genome 3D topology in these cells, respectively.
Founding:
INRAE, Animal Genetics division, 2015-2017
Project status:
Phase 1 completed, Phase 2 ongoing
Main collaborators:
INRAE
Role in this project:
Co-coordinator, leader of the data analysis WP
HiC-DOC
 HiC-DOC: detecting and comparing genomic compartments from Hi-C data
HiC-DOC: detecting and comparing genomic compartments from Hi-C data
Project description:
Genomic compartmentalization is a biological factor affecting cell functionality. Different compartments
can be observed in the nucleus of eukaryotic cells, grouping genomic regions into clusters. Active
compartments are usually associated with open chromatin and gene expression while inactive compartments
are usually associated with closed chromatin and gene repression [1]. Analysis of data produced by the Hi-C
protocol reveals compartmentalization of chromatin in the nucleus, which can vary as a tissue develops.
Today, existing methods to detect genomic compartmentalization are limited in at least one of the following
ways: detecting compartments qualitatively with no confidence measure, ignoring experimental biases,
and/or dismissing replicate variability.
We propose an improvement over existing methodology to detect compartments and compare
compartmentalization between conditions. First, we properly correct the diverse biases inherent to Hi-C data,
using cyclic loess normalization [2] to reduce technical biases and Knight-Ruiz matrix balancing [3] to
mitigate biological biases. Then, we correct interaction counts with a loess regression to clearly expose the
compartmentalization information captured by the data. Finally, we use an unsupervised learning method,
constrained K-means [4], to computationally detect compartments from the normalized data. This method
enables us to produce quantitative “concordance” values for each genomic region in each replicate,
supporting our compartment predictions. Finally, we use these concordance values for differential analysis of
compartmentalization between conditions. From their distributions, we obtain p-values revealing the
significance of each predicted compartment change.
Founding:
INRAE
Project status:
Ongoing
Main collaborators:
INRAE (MIAT)
Role in this project:
Co-coordinator, contribution to the bioinformatics analysis
TREE-DIFF
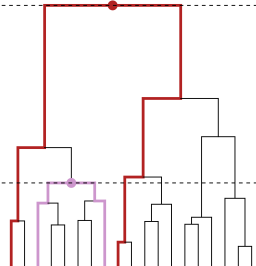 Tree-diff: detecting significant changes between groups of trees from HAC
Tree-diff: detecting significant changes between groups of trees from HAC
Project description:
Trees are frequently used to describe data that are organized hierarchically. This
is the case, for instance, in phylogeny, or when using statistical methods such as
hierarchical agglomeration clustering (HAC) or Classification and Regression Trees.
The TREE-DIFF projects aims at developing an approach to provide statistical
guarantees for the comparison between two sets of trees corresponding to two different
conditions. Our approach builds on tree distances and an aggregation procedure for
moderated Student tests performed at the level of leaf pairs. Numerical experiments
confirm its statistical validity even for small sample size. The method is illustrated
with various practical applications in the field of biology, including Genome Wide
Association Studies (GWAS), study of chromatin conformation with Hi-C data and
phylogenetic studies.
Founding:
INRAE, CNRS
Project status:
Ongoing
Main collaborators:
INRAE (MIAT), CNRS (IMT), INRIA
Role in this project:
Partner, contribution to the bioinformatics analysis
Plus4Pigs
Getting true Pluripotent Stem Cells in Pigs: a key step for large scale ex-vivo “Genotype to Phenotype” studies
Project description:
Current global changes (global warming, availability of agricultural resources, societal perception of animal husbandry, health importance of zoonoses) are forcing us to rethink our production systems. The pressure of animal production on ecosystems must be reduced, food and health security must be increased and animal welfare in breeding must be better addressed. To achieve these global objectives, the integration of the digital dimension for the management of farms is essential. Its coupling with innovative cellular systems will make it possible to evaluate at high-speed phenotypes that are difficult to measure in breeding on live animals, and therefore to acquire quantities of data suited to the methodologies developed for «big data«. This strategy also reduces the need for animal testing in accordance with the 3R rule (Replace, Reduce, Refine). We propose, within the framework of this project, to use the numerical dimension from multi-omics data at the single-cell and tissue scale to predict the molecules necessary and sufficient to maintain porcine pluripotency and transfer this knowledge for the production and use of porcine pluripotent stem cell lines (PSCs) for animal and human health applications. This approach, breaking with traditional experimental approaches, will represent a major breakthrough for genetic, pharmaceutical or toxicological studies. Indeed, improving resistance to animal diseases has long been a research priority that is struggling to progress due to the lack of high throughput phenotyping method. The objectives of the project are as follows: 1) Molecular characterization of the microenvironment of the porcine embryo before implantation. 2) Production of cell lines with reporter systems allowing tracing and sorting of porcine pluripotent cells. 3) Optimization of the combination of exogenous factors necessary for reprogramming to a state of authentic pluripotency. 4) Identification of signaling molecules necessary and sufficient for the maintenance of porcine pluripotent cells in vitro. 5) The production of porcine pluripotent lines with full potential for differentiation
Founding:
ANR, France
Project status:
Ongoing
Main collaborators:
INRAE
Role in this project:
Contribution to the bioinformatics analysis
Pig3Dgenome
 Pig3Dgenome: comparing the 3D genomic structure of porcine muscle cells during late development
Pig3Dgenome: comparing the 3D genomic structure of porcine muscle cells during late development
Project description:
The three dimensional organization of the genome plays a major role in the regulation of gene expression. Chromosome territories, compartments, topological domains, and loops, are the main features of the genome topology. Most of these features are quite stable ensuring a suitable niche for maintaining either transcriptional activation or repression. However, the structural plasticity of the chromatin also permits conformational changes that may lead to alterations in the transcriptional activity. These dynamic changes are particularly remarkable during gene expression reprograming occurring in early development (i.e. zygote genome activation, transition from pluripotent to lineage-committed cells, and cell differentiation). However, these dynamic events remain poorly understood, especially those concerning late development and tissue maturity processes. Our study offers new insights into the 3D genome organization dynamics at late gestation in mammals. More precisely, we addressed the global genome organization of porcine muscle nuclei at 90 and 110 days of gestation by performing in situ Hi-C experiments. This stage of gestation is a relevant period for porcine muscle development and maturity, as already shown in a previous transcriptome study. We obtained evidence of important topological changes in the 3D genome structure at this period that are associated to variations in gene expression. This dynamic changes correspond to a global fragmentation of the genome, switches of compartment type, differential chromatin interactions and dynamics of the telomeric regions.
Founding:
INRAE, CNRS
Project status:
Completed
Main collaborators:
INRAE (MIAT), CNRS (IMT)
Role in this project:
Coordinator, leader of the bioinformatics analysis
ENCODE
 ENCODE: ENCyclopedia Of Dna Elements
ENCODE: ENCyclopedia Of Dna Elements
Project description:
The Encyclopedia of DNA Elements (ENCODE) Consortium is an ongoing international collaboration of research groups funded by the National Human Genome Research Institute (NHGRI). The goal of ENCODE is to build a comprehensive parts list of functional elements in the human genome, including elements that act at the protein and RNA levels, and regulatory elements that control cells and circumstances in which a gene is active.
ENCODE investigators employ a variety of assays and methods to identify functional elements. The discovery and annotation of gene elements is accomplished primarily by sequencing a diverse range of RNA sources, comparative genomics, integrative bioinformatic methods, and human curation. Regulatory elements are typically investigated through DNA hypersensitivity assays, assays of DNA methylation, and immunoprecipitation (IP) of proteins that interact with DNA and RNA, i.e., modified histones, transcription factors, chromatin regulators, and RNA-binding proteins, followed by sequencing.
Founding:
NHGRI
Project status:
Ongoing but my personal contribution was mainly during the early phases of the project (pilot phase and ENCODE2)
Main collaborators:
CRG, Affymetrix, CSHL, UCSC
Role in this project:
Contributions to the bioinformatics analysis