Input data and parameters
QualiMap command line
| qualimap bamqc -bam SRR7062654_1.bam -c -nw 400 -hm 3 -sd |
Alignment
| Command line: | bwa mem -K 100000000 -R @RG\tID:1\tPU:1\tSM:SRR7062654\tLB:SRR7062654\tPL:illumina -t 10 -M Gallus_gallus.Gallus_gallus-5.0.dna.toplevel_chr25-26.fa SRR7062654_R1.fastq.gz SRR7062654_R2.fastq.gz |
| Draw chromosome limits: | yes |
| Analyze overlapping paired-end reads: | no |
| Program: | bwa (0.7.17-r1188) |
| Analysis date: | Mon Oct 25 12:08:08 UTC 2021 |
| Size of a homopolymer: | 3 |
| Skip duplicate alignments: | yes (only flagged) |
| Number of windows: | 400 |
| BAM file: | SRR7062654_1.bam |
Summary
Warnings
| No flagged duplicates are detected | Make sure duplicate alignments are flagged in the BAM file or apply a different skip duplicates mode. |
Globals
| Reference size | 8,220,070 |
| Number of reads | 1,987,908 |
| Mapped reads | 1,986,265 / 99.92% |
| Unmapped reads | 1,643 / 0.08% |
| Mapped paired reads | 1,986,265 / 99.92% |
| Mapped reads, first in pair | 993,166 / 49.96% |
| Mapped reads, second in pair | 993,099 / 49.96% |
| Mapped reads, both in pair | 1,984,622 / 99.83% |
| Mapped reads, singletons | 1,643 / 0.08% |
| Secondary alignments | 698 |
| Read min/max/mean length | 30 / 90 / 90.01 |
| Duplicated reads (flagged) | 0 / 0% |
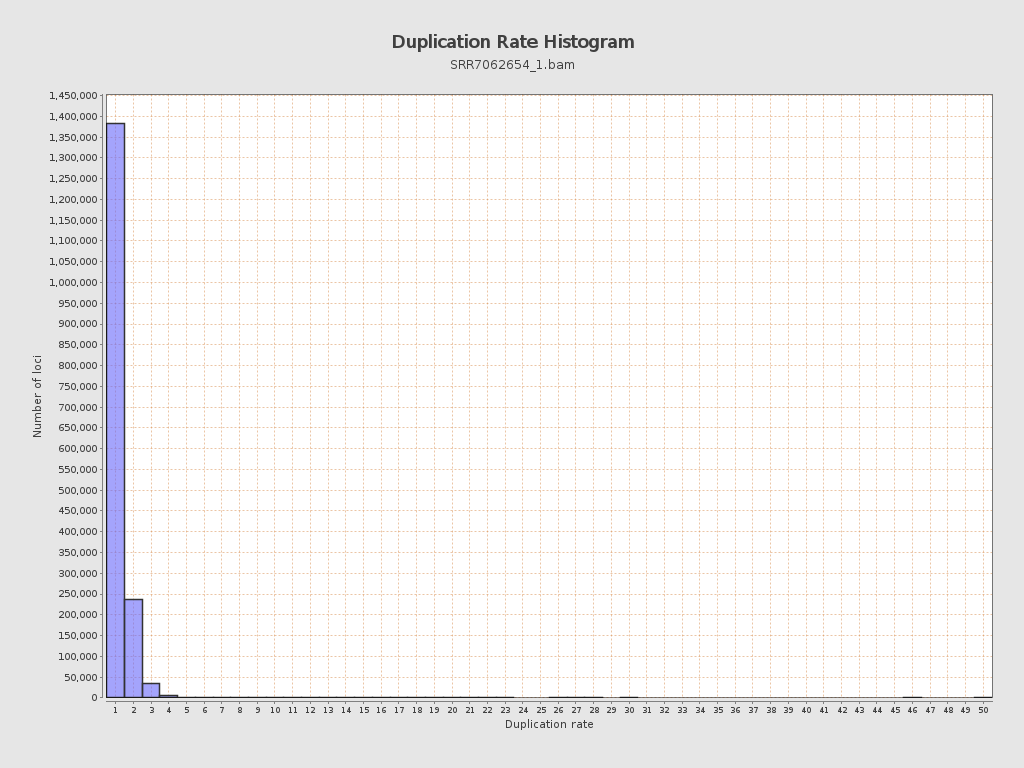
| Duplicated reads (estimated) | 327,476 / 16.47% |
| Duplication rate | 16.66% |
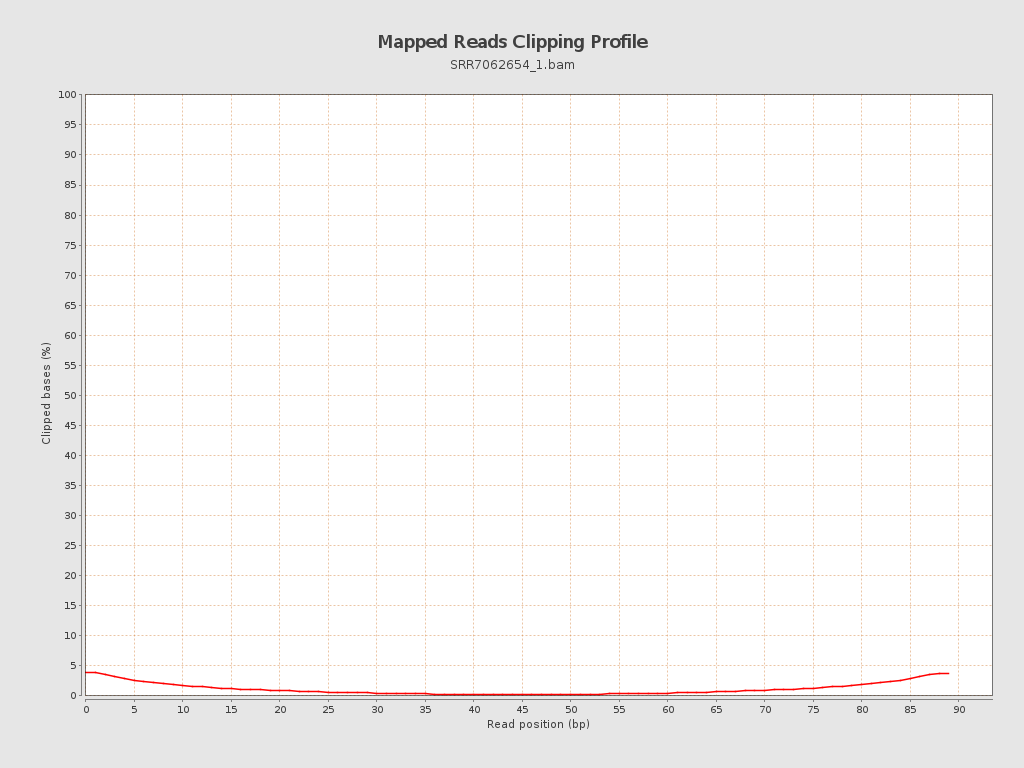
| Clipped reads | 59,419 / 2.99% |
ACGT Content
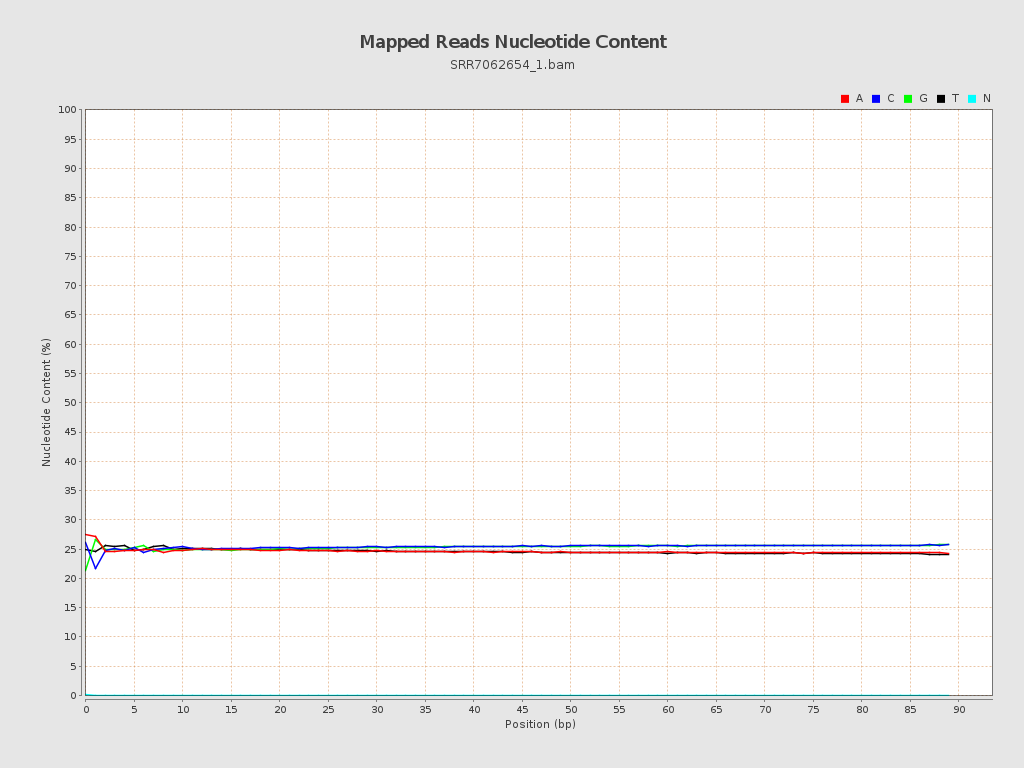
| Number/percentage of A's | 43,813,202 / 24.63% |
| Number/percentage of C's | 45,163,359 / 25.39% |
| Number/percentage of T's | 43,752,660 / 24.6% |
| Number/percentage of G's | 45,092,509 / 25.35% |
| Number/percentage of N's | 32,795 / 0.02% |
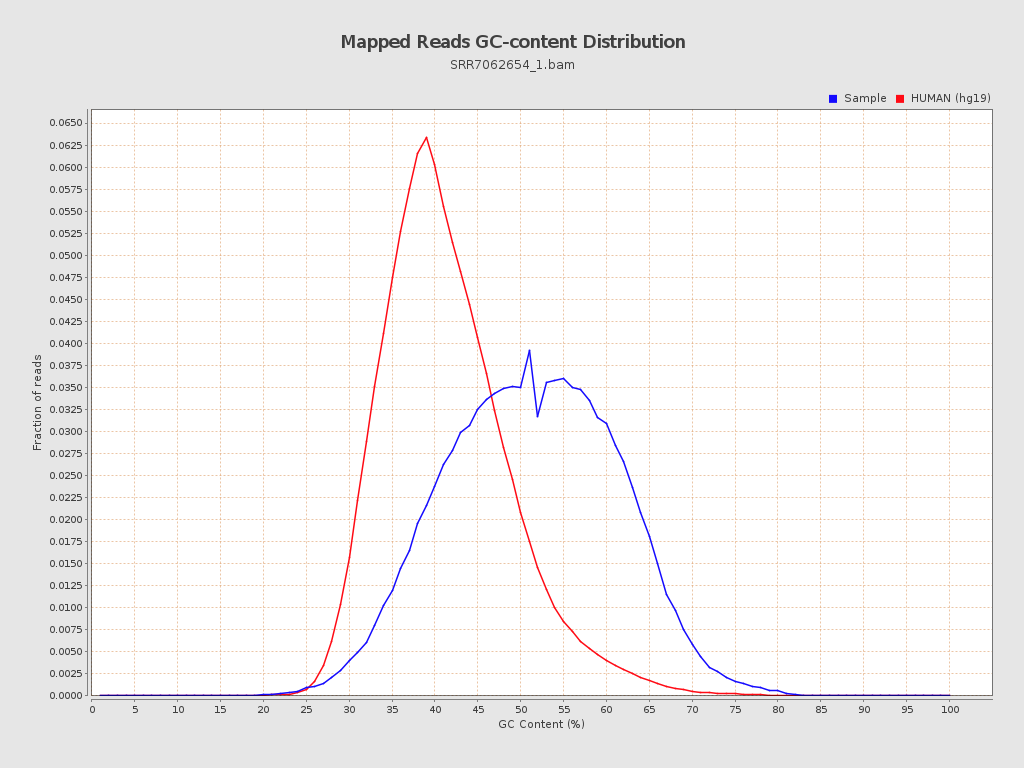
| GC Percentage | 50.75% |
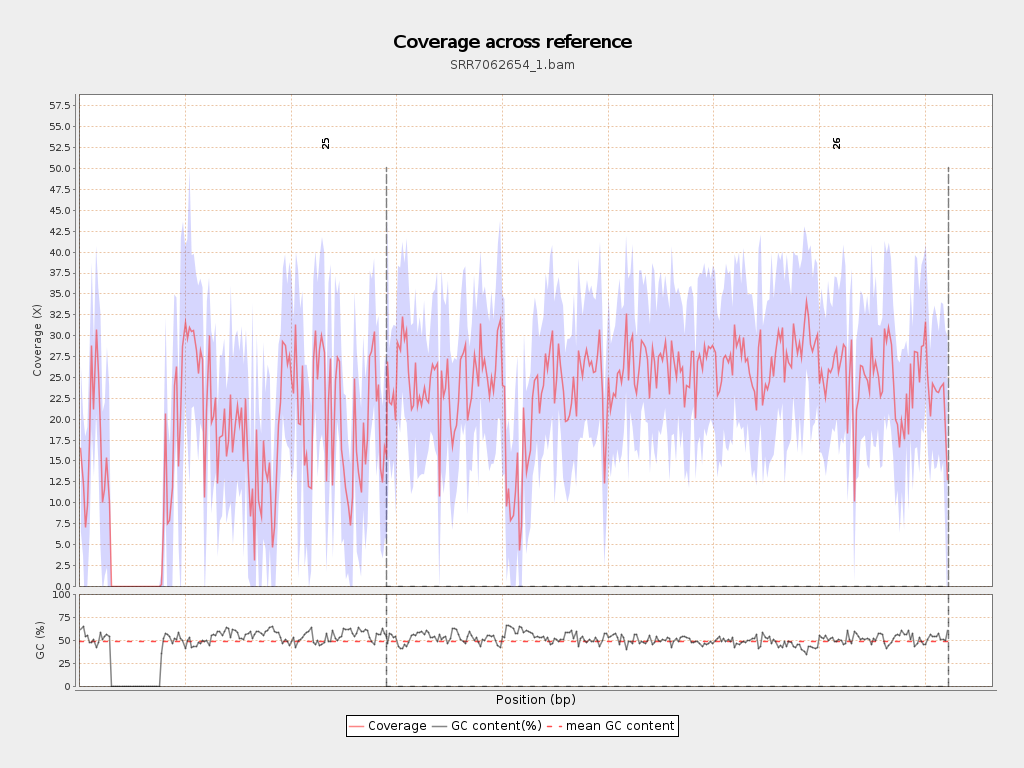
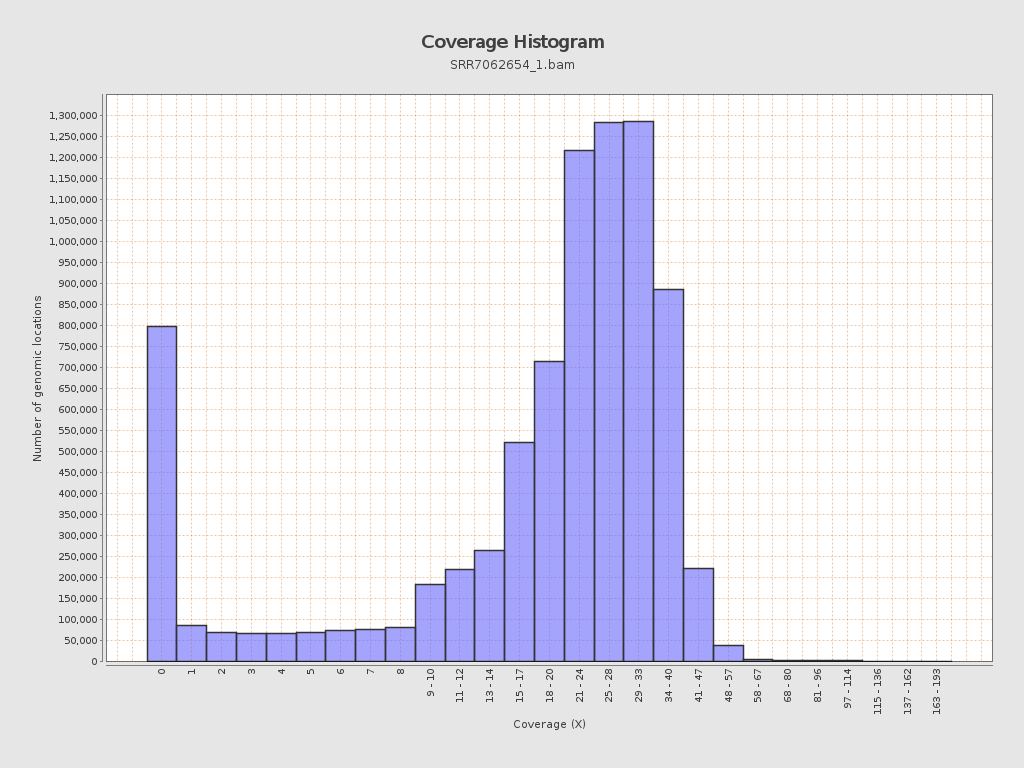
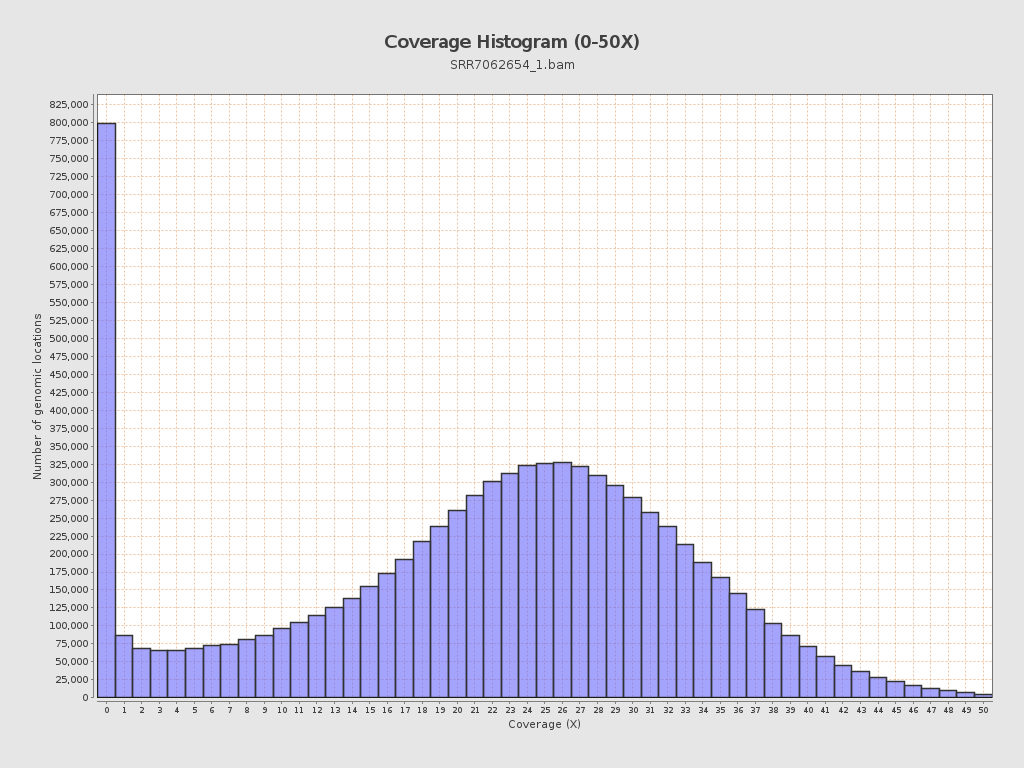
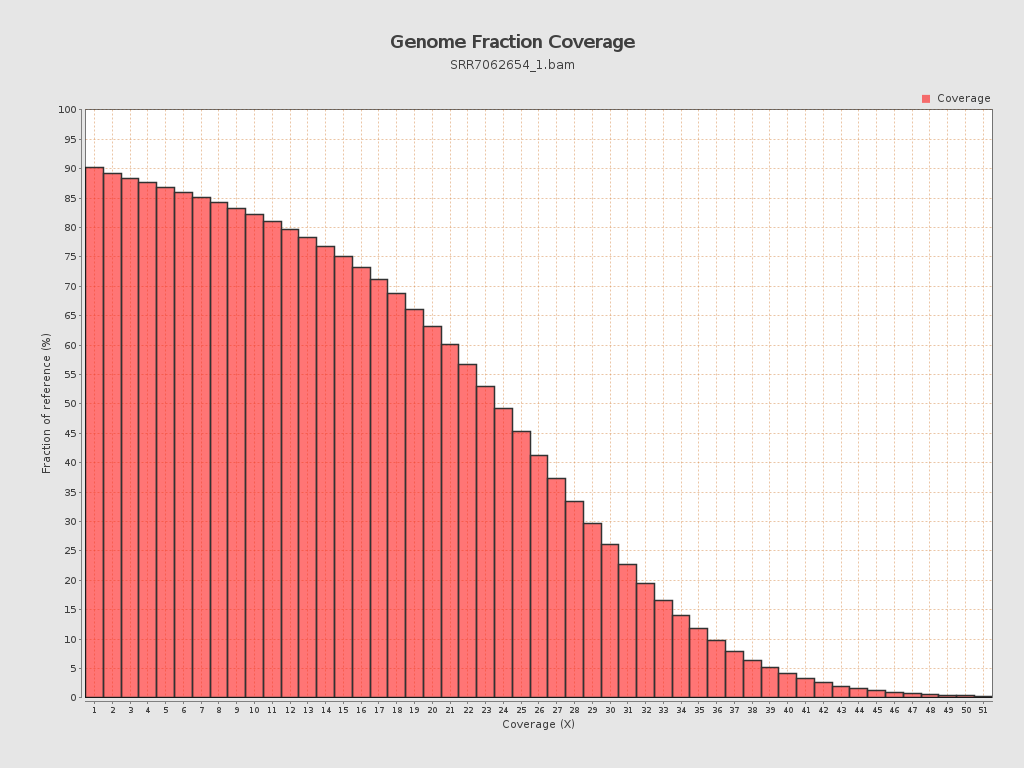
Coverage
| Mean | 21.65 |
| Standard Deviation | 11.8152 |
Mapping Quality
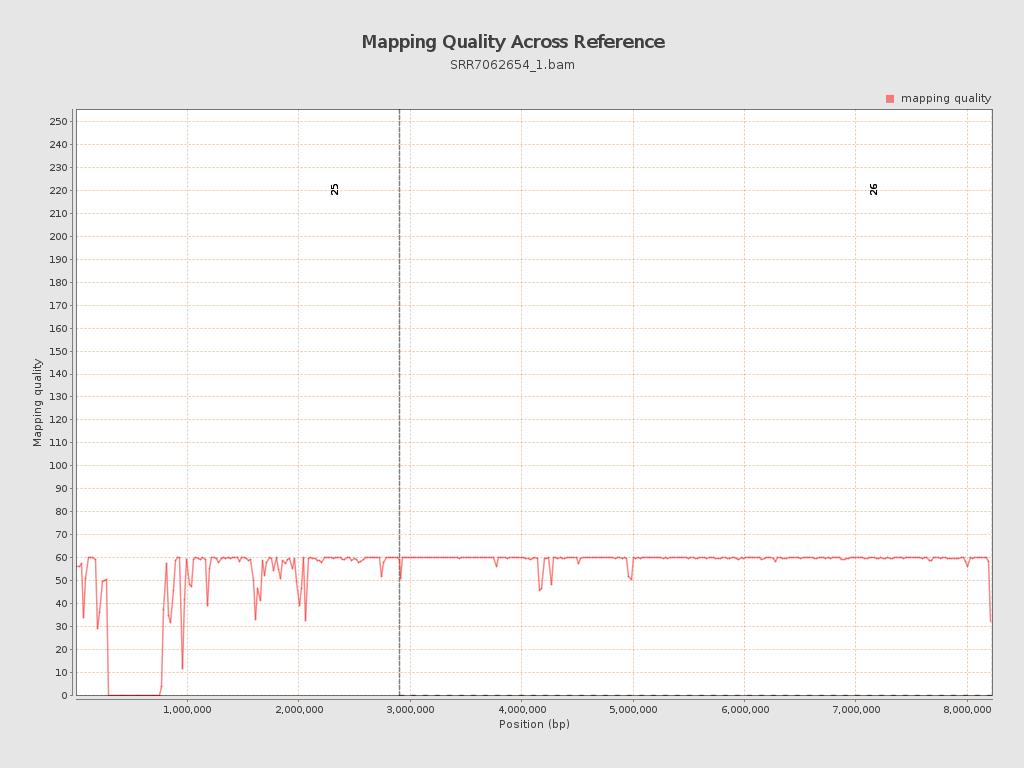
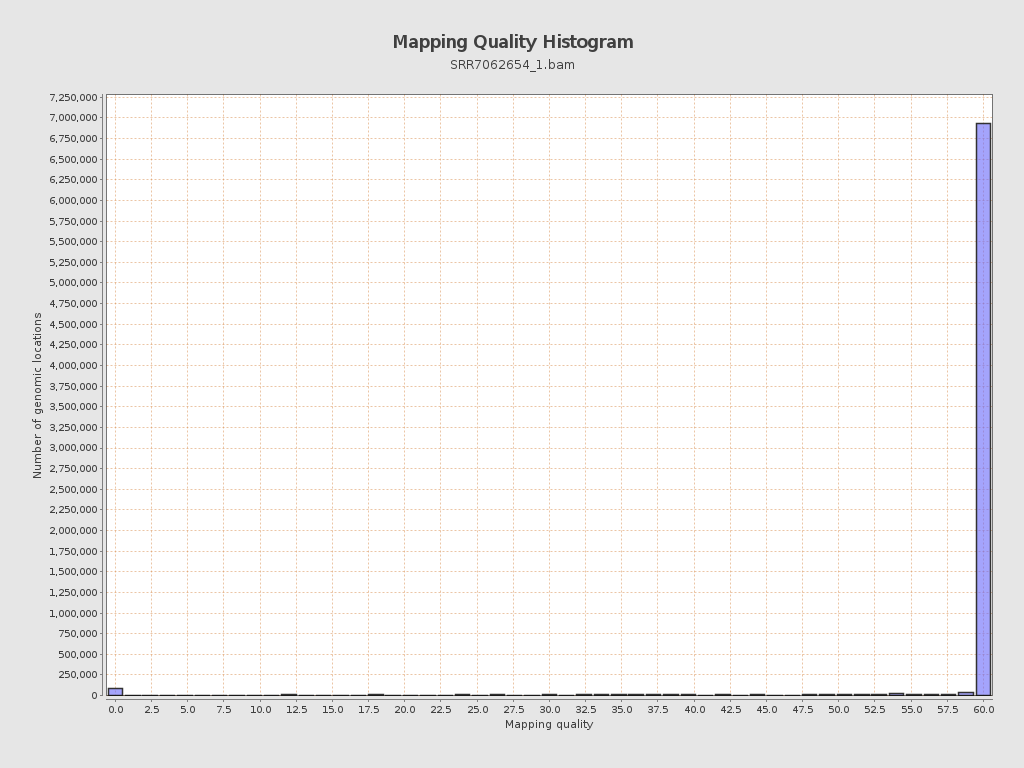
| Mean Mapping Quality | 54.69 |
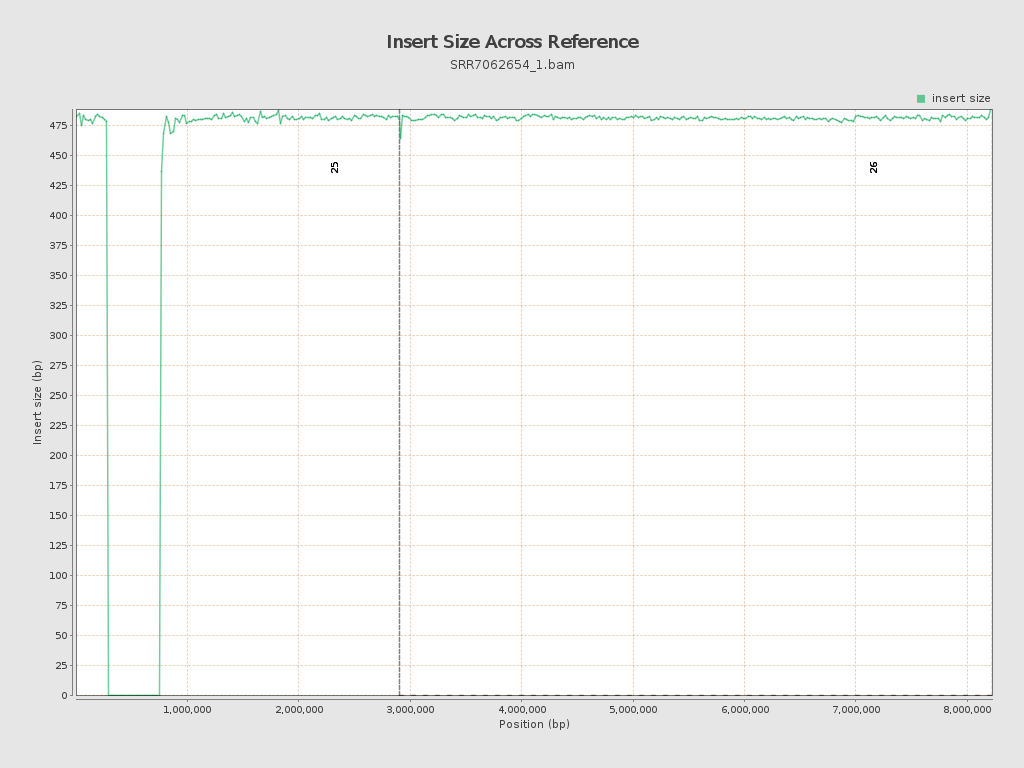
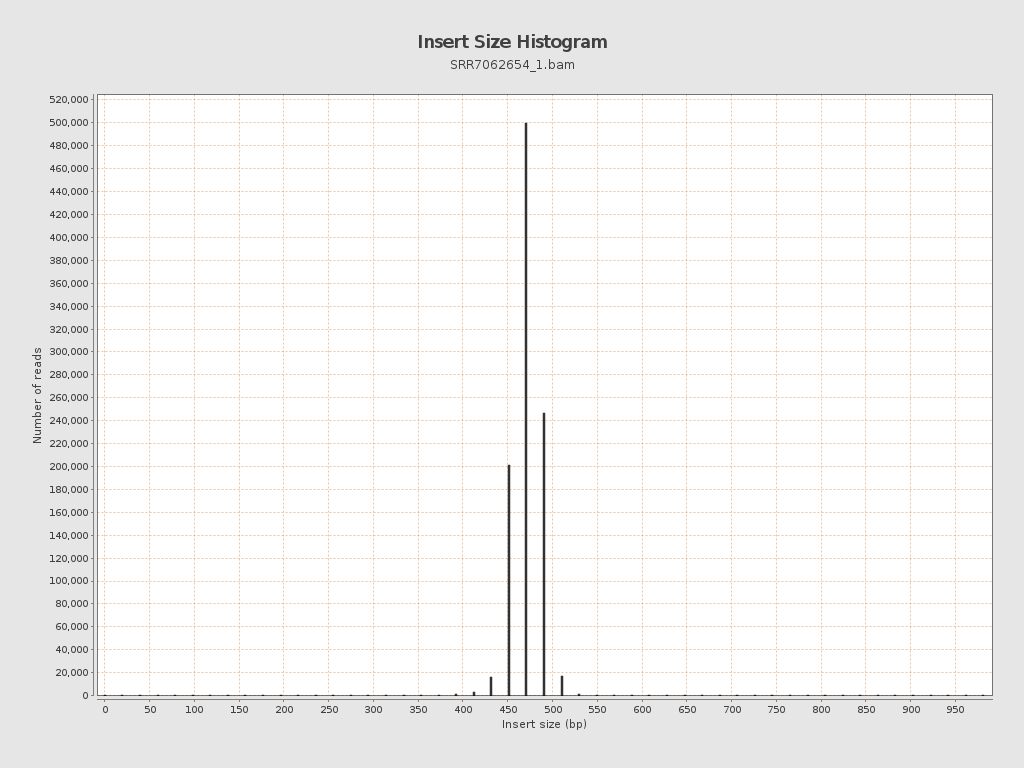
Insert size
| Mean | 586.97 |
| Standard Deviation | 11,397.04 |
| P25/Median/P75 | 472 / 481 / 491 |
Mismatches and indels
| General error rate | 1% |
| Mismatches | 1,698,916 |
| Insertions | 34,815 |
| Mapped reads with at least one insertion | 1.71% |
| Deletions | 39,461 |
| Mapped reads with at least one deletion | 1.9% |
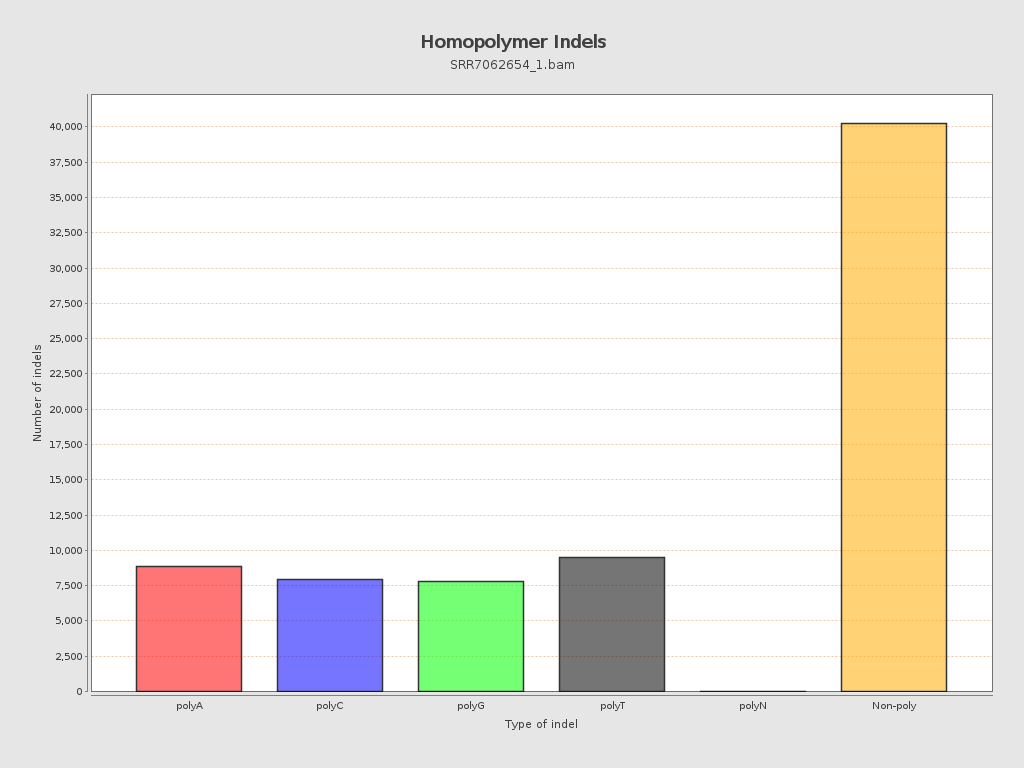
| Homopolymer indels | 45.8% |
Chromosome stats
| Name | Length | Mapped bases | Mean coverage | Standard deviation |
| 25 | 2906300 | 46291538 | 15.928 | 13.4294 |
| 26 | 5313770 | 131673369 | 24.7797 | 9.4671 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}